| Genetic Algorithms Reverse Resistance To Antibiotics |
| Written by Mike James | |||
| Wednesday, 13 May 2015 | |||
|
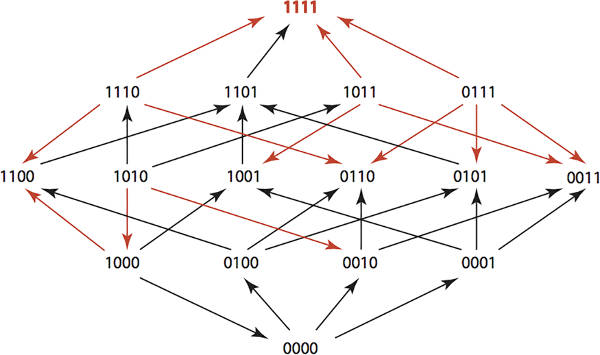
If you use antibiotics against bacteria then antibiotic resistance is inevitable. However, if you use multiple antibiotics in a clever way you can engineer the bacterial population to remain unchanged. This algorithmic approach to controlling evolution is one way that we might retain the usefulness of antibiotics for generations to come and also has wider applications. If you use a single antibiotic against a population of bacteria then you will kill most or all of the organisms that are susceptible to it. The few bacteria left alive form the start of a new population resistant to the antibiotic. This is evolution in action. As the population is subjected to a pressure the fittest organisms survive and increase as a proportion of the whole. Notice that this is going to happen even if antibiotics aren't overused. Overuse speeds the process, but being restrained in antibiotic use still leads to resistance eventually. Annually more than 2 million people in the United States get infections that are resistant to antibiotics and at least 23,000 people die as a result, according to the CDC, Centers for Disease Control and Prevention. One approach to using antibiotics to avoid selection for resistance is to rotate the antibiotics used. In this way you hope to avoid applying a single evolutionary pressure to make the population move in the direction of resistance to any specific or multiple antibiotics. Taking this blind rotation to a more sophisticated level, what if we could design optimal antibiotic use schedules that would maintain a population in its original state and even return a resistant population back to the wild state? Miriam Barlow of the University of California, Merced, and mathematician Kristina Crona of American University of Washington, DC have done just this - and the algorithm is interesting and is possibly capable of improvement. Keeping the biology simple means we can concentrate on the algorithm. If you want to know the details see the paper on the Plos One website. The study considered four possible amino acid substitutions, each of which confer some resistance to antibiotics. We can identify a genotype as a four-bit number with 0000 being the wild type and a 1 indicating a substitution. If we assume that the probability of substitution is low then we can assume that only one change occurs at any given time. This means that the genotypes form a simple Hamming Graph. That is, we have a graph with 16 nodes and connections between nodes that differ by one bit - i.e. they are one Hamming distance apart. You can measure the evolutionary pressure of any given antibiotic by measuring the tendency for any genotype to decrease in favour of a genotype that differs by one amino acid. For example, for Ampicillin we have the following Hamming graph:
The red arrows indicate growth rate differences that are significant. You can think of the red arrows as showing evolutionary changes that are likely due to the application of the antibiotic. You can also arrange this data into a 2 x 2 x 2 x 2 tensor. i.e. a 4-dimensional array giving the fitness landscape for the genotypes under a given antibiotic. The study used 15 antibiotics so there are 15 fitness tensors. If you administer a sequence of antibiotics then the overall fitness landscape is given by the composition of their tensors. The problem is to find an optimal sequence of k antibiotics such that the composed fitness tensor C maximises the probability of moving from any genotype u to the wild type 0000.
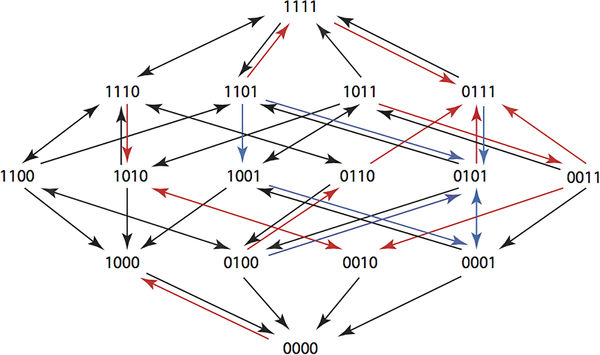
A composite Hamming graph for a 6 step plan showing paths from 1111 to 0000. You can see that in the new fitness landscape arrows point back to the wild type. This requires searching all 15k sequences of length k. It is big problem, but one that can be tackled for k up to 6 using complete enumeration in a Maple program. The key observation, if you want to get involved is: "The running time of the program is slow because of the exponential growth in the number of sequences. At present we do not know whether an efficient algorithm exists for solving our optimization problem for larger values of k." Two types of optimization were tried. The first just used the arrow directions and the second used the actual growth rates. Using the actual growth rates gave better results and it was possible to find six step plans that returned any genotype to the wild state with probabilities at least 0.6. The point here is that these plans will return a resistant population back to the wild state and then keep the wild state intact while allowing the physician to use antibiotics to kill members of the population. The researchers plan to try the treatment plans in a clinical setting in the future and also work on better optimization tools. There are other obvious possibilities in using directed evolution in this way. Perhaps we can avoid breeding resistant weeds and insects by rotating treatments. There might also be ways of returning ecosystems to the wild state by applying an optimal cyclic treatment - a sort of super crop rotation.
"Disc method for identifying sensitivity" by Netha Hussain
This could well be the genetic algorithm that makes a difference. More InformationRelated ArticlesA New DNA Sequence Search - Compressive Genomics Speed Data Is Enough To Track You How Algorithms Changed The World NYT on the Future of Computing Pancake flipping is hard - NP hard
To be informed about new articles on I Programmer, install the I Programmer Toolbar, subscribe to the RSS feed, follow us on, Twitter, Facebook, Google+ or Linkedin, or sign up for our weekly newsletter.

Comments
or email your comment to: comments@i-programmer.info
|
|||
| Last Updated ( Thursday, 14 May 2015 ) |